
【2014諾貝爾化學獎深入報導】 打破光學顯微鏡的解析度極限-超高解析螢光顯微法

【2014諾貝爾化學獎深入報導】 打破光學顯微鏡的解析度極限:超高解析螢光顯微法
東京大學化學所專案助理教授邱亮達 撰文/東京大學理學博士陳藹然 責任編輯
今年的諾貝爾化學獎頒給了由 Stefan Hell, William Moerner 和 Eric Betzig 所開發的超高解析螢光顯微法。所謂的超高解析螢光顯微法,就是能打破光的繞射極限(圖一)的顯微鏡技術。
得獎的顯微法有兩種:受激放射消去顯微法STED (STimulated Emission Depletion) Microscopy和光啟動定位顯微法PALM (PhotoActivated Localisation Microscopy)。這兩種方法是完全獨立被開發出來的方法,現今多應用在生物研究上。
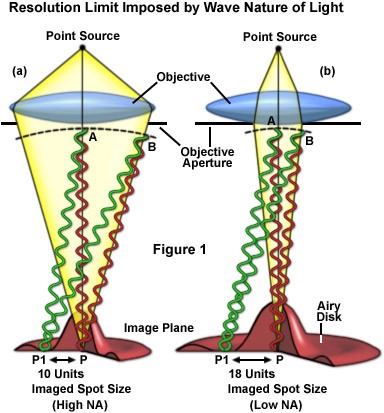
圖一
圖片來源:
http://www.microscopyu.com/articles/superresolution/diffractionbarrier.html
【圖一】假設由樣本發出的光視同於由點光源(Point Source)發出的光,當這些光線經過顯微鏡物鏡時會與物鏡孔洞(Objective Aperture)的邊緣產生繞射。這些繞射的光在共軛面上會產生相長干涉(圖中紅線)及相消干涉(圖中綠線),造成呈像面(Image Plane)面上的影像不再是一個點而是一個被稱為光暈盤(Airy Disc)的干涉圖樣 。這也是為什麼光的聚焦永遠無法小於其波長除以2倍數值孔徑大小。圖a與圖b則清楚呈現了不同的物鏡數值孔徑(NA)如何影響呈像面上干涉圖樣(意即光學解析度)的大小。
以下將簡略地介紹為這三人摘下諾貝爾獎桂冠的研究。技術與理論的細節會放在各圖的圖解中,有興趣者請細讀圖解。
受激放射消去顯微法: STED (STimulated Emission Depletion) Microscopy
此技術由理論1,2到實做3的基礎都是由 Stefan Hell 所確立的。這三篇論文也為他贏得了這次諾貝爾化學獎的殊榮。「受激放射消去顯微法」採用與一般高解析度的共軛焦顯微鏡類似的聚焦掃瞄呈像方式。這個技術的核心概念是:假設一道光的聚焦永遠無法突破繞射極限,那就用兩道不同的光令其與螢光標誌交互作用而達成超高解析度的目標。
這兩道光中,其中一道以平常手法聚焦的激發光用以激發螢光分子、另一道聚焦為甜甜圈圖案的抑制光則用以抑制除了甜甜圈中心之外所有被激發的螢光分子發光(圖二)。
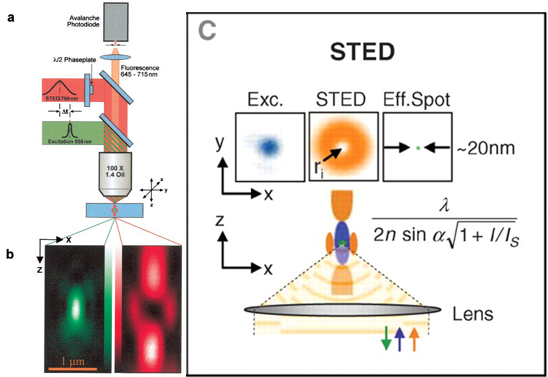
圖二 (圖片來源:圖a,b取自參考文獻3, 圖c取自參考文獻4)
【圖二】受激放射消去顯微法的運作原理。圖a是受激放射消去顯微法的實驗裝置圖解。綠色路徑為激發螢光色素用的光路(Excitation)、紅色路徑則為用以抑制在外圍的被激發螢光分子發光的光路(STED)。圖b為激發光(綠)及抑制光(紅)在焦點平面上於XZ平面的圖示。為了產生圖b中紅光的中空焦點,圖a紅光路徑的半波片(λ/2 Phaseplate)中心有鍍上一層光遮罩。圖c中上列的圖示是說明受激放射消去顯微法如何增強XY平面上的光學解析度。下列圖示則是說明其如何增強XZ平面上的光學解析度。此圖中的藍光為激發光(Exc.)、橘光為抑制光(STED)、綠光則為實際可偵測到的螢光(Eff. Spot)。圖中公式為受激放射消去顯微法理論上的解析度極限。分母中的 I 為實際入射所使用的抑制光的強度,Is為成功抑制螢光所需的抑制光強度。也就是說,若實際入射光無限大,則此系統的所能解析的物質大小則趨近於零,意即解析度無限大。
當甜甜圈圖案中心的孔洞小於繞射極限時,也就是說只有小於繞射極限範圍的螢光分子能夠正常發光時,其呈現地影像將超越繞射極限的影像解析度。至於抑制被激發螢光分子發光的方法,在Stefan Hell初期的研究裡是利用受激放射消去的原理(圖三),也是這個方法得名的由來。由於這個手法的理論解析度趨近於無限大(圖二公式),並且理論上可以適用於任何已知的螢光標誌上,因此發表後很快地便獲得了極大的注目。近年來,為了解決這個手法一直以來最被垢病的問題:強烈的抑制光所造成的樣本損傷;許多使用不同螢光消去原理的同類型裝置紛紛被開發4(主要著力在可用微弱光線即關閉其發光機制的新型螢光標誌分子的研究),也讓人十分期待這個技術未來能有如何的應用及發展。
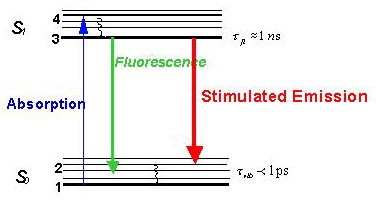
圖三(圖片來源:圖取自Stefan Hell實驗室網頁http://www3.mpibpc.mpg.de/groups/hell/STED.htm)
【圖三】受激放射消去的原理。一個螢光物質在吸收(Absorption)了特定高能波長的光線後,其原本位於S0電子基態的分子構造會被激發到S1第一電子激發態。此時若什麼都不做,其能階將會釋放使其分子構造回到S0電子基態,同時發出螢光(Fluorescence)。不過在電子態被激發時,若同時打入一道受激放射光,則原本位於S1第一電子激發態的分子會受激放射(Stimulated Emission),即釋出與受激放射光同樣能量的光線並回到S0電子基態的某個特定振動能階(此原理與雷射發光是相同的)。此時若觀察與受激放射光不同波長的螢光放射區段則無法看到螢光發光,因而達到抑制發光的效果。
光啟動定位顯微法:PALM (PhotoActivated Localisation Microscopy)
「光啟動定位顯微法」是一種一次觀測單一螢光分子的顯微法。此技術的理論5及實做6都由 Eric Betzig 完成。不過由於此類單分子觀測技術是由William Moerner開先河7,再加上他對單一綠色螢光蛋白分子的研究直接影響了「光啟動定位顯微法」的實踐8,因此這四篇論文讓這兩人與Stefan Hell共同分享了今年的諾貝爾化學獎。此技術直接放棄聚焦掃瞄的呈像方式,改用全面照明的系統架構。此呈像手法的關鍵在於須要被光啟動之後才會發光的螢光標誌分子。其運作原理是:既然已知聚焦光無法分辨相距在繞射極限範圍內的兩個螢光標誌,那就改利用光啟動的方法讓相距在繞射極限範圍內的螢光標誌分開發光,之後逐一啟動及消去各個螢光標誌的發光能力,並一一標記各發光過的螢光標誌的位置,最後就能組成一個以單一發光分子為解析度的螢光圖像9(圖四)。早期,由於這個呈像方法需要不段重覆地點亮螢光標誌分子並待其光致褪色,此技術最為人所垢病地就是過慢的呈像速度以致其只能被應用於被固定的細胞上。為了改善這個窘境,許多科學家紛紛投入研究新式的可逆式光轉換螢光分子(也就是可以光啟動也可以光關閉的螢光分子),也使得光啟動定位顯微法的呈像速度越來越快,現在甚至已可用來觀測活細胞中單一被標誌分子的動態10。

圖四 (圖片來源:圖取自參考文獻9)
【圖四】光啟動定位顯微法的運作原理。現假設有一個間隔遠小於繞射極限、並由許多螢光標誌分子所標記、如同右上角圖t的橘色同心圓圖樣的圖形。由於其圓與圓之間的間隔遠小於繞射極限,此時若是用一般的光學顯微法觀測此樣本,其呈現出來的影像會是同於圖s般的白色圖樣般的圓盤。要以光啟動定位顯微法觀測此圖形的細微構造,其前題是必須要使用特殊、須要被光啟動後才能發光的螢光標誌分子標記此圖形。首先,對被此種特殊螢光標誌所標記的樣本開啟激發螢光標誌分子用的激發光。這道激發光從頭到尾都會保持持續開啟的狀態。此時由於尚未有任何螢光標誌分子被啟動,因此視野內不會有任何螢光發出(圖a, b)。
接下來,用非常微弱的啟動光照射這個樣本。這個啟動光必須要弱到在成千上萬的特殊螢光標誌中,只有少數的一兩個會被隨機啟動。啟動光的照射在此圖中以紫燈表示。此時由於有極少數的特殊螢光標誌被啟動了,在視野中可以看到零散地螢光分子發光了(圖c)。在以高斯分佈計算圖c中每個光暈盤的中心位置之後,將其定位標記下來,製成如圖d的圖。接下來等圖c中被激發的螢光分子被光致褪色(螢光分子被持續地激發所導致地永久性發光機制受損的現象總稱)後,視野再度恢復如圖e般暗淡。
不過由於被光致褪色的螢光標誌的定位已被紀錄,圖f仍保留的圖d留下來的定位。之後再度照射啟動光,又有其它尚未被光致褪色的螢光標誌被激發,視野再度呈現如圖g的圖樣。將這些光暈盤再度定位後加到之前的定位紀錄上,就會看到如圖h、包含之前和此次測定所得到的定位標記的圖。等這一輪的螢光標誌分子也被光致褪色後(圖i),再度照射啟動光,並持續紀錄每輪所能標記到的定位,其定位標記圖就會如圖j, n, r一般漸漸呈現受標記圖形的輪廓。最後當每一個分子都被光啟動定位後,其全部的定位標記相加就能得到被標記樣本的超高解析度影像(圖t)。
由以上的介紹,大概不難瞭解這兩個超高解析螢光顯微法現在的突破都要仰賴新式螢光分子的開發。這個由物理學家起頭(Stefan Hell和Eric Betzig的本行都算是物理學者)的新領域現在要靠化學家做進一步突破,以期將來能夠解決更多生物上的難題。如此跨學門的研究方向實屬少見,也再再突顯出此一新技術受到矚目的程度!
參考文獻
1. Hell, S. W. & Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780 (1994).
2. Hell, S. & Kroug, M. Ground-state-depletion fluorscence microscopy: A concept for breaking the diffraction resolution limit. Appl. Phys. B 60, 495–497 (1995).
3. Klar, T. a., Jakobs, S., Dyba, M., Egner, a. & Hell, S. W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. 97, 8206–8210 (2000).
4. Hell, S. W. Far-field optical nanoscopy. Science 316, 1153–8 (2007).
5. Betzig, E. Proposed method for molecular optical imaging. Opt. Lett. 20, 237 (1995).
6. Betzig, E. et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–5 (2006).
7. Moerner, W. & Kador, L. Optical detection and spectroscopy of single molecules in a solid. Phys. Rev. Lett. 62, 2535–2538 (1989).
8. Dickson, R., Cubitt, A., Tsien, R. & Moerner, W. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 388, 355–358 (1997).
9. Gould, T. J., Verkhusha, V. V & Hess, S. T. Imaging biological structures with fluorescence photoactivation localization microscopy. Nat. Protoc. 4, 291–308 (2009).
10. Sengupta, P., van Engelenburg, S. B. & Lippincott-Schwartz, J. Superresolution imaging of biological systems using photoactivated localization microscopy. Chem. Rev. 114, 3189–202 (2014).
 前一篇文章
前一篇文章 下一篇文章
下一篇文章![[影音] CASE【百秒說科學】《改變時空形狀的重力波》](https://highscope.ch.ntu.edu.tw/wordpress/wp-content/uploads/2016/12/Online-620x280.jpg) [影音] CASE【百秒說科學】《改變時空形狀的重力波》
[影音] CASE【百秒說科學】《改變時空形狀的重力波》  2017 天文大事紀
2017 天文大事紀 ![[影音] CASE【百秒說科學】《交叉分子束》](https://highscope.ch.ntu.edu.tw/wordpress/wp-content/uploads/2016/11/CMB-620x280-科學Online.jpg) [影音] CASE【百秒說科學】《交叉分子束》
[影音] CASE【百秒說科學】《交叉分子束》  悲傷與快樂的音樂引起不同的腦部活化型態
悲傷與快樂的音樂引起不同的腦部活化型態  發現比地球大一點又老一點的親戚
發現比地球大一點又老一點的親戚  【2019諾貝爾化學獎】鋰離子電池
【2019諾貝爾化學獎】鋰離子電池  棉籽粕中殘留的棉籽酚
棉籽粕中殘留的棉籽酚  伊波拉病毒(Ebola virus)—病毒的防制
伊波拉病毒(Ebola virus)—病毒的防制 


Nobel Prize
沒有看不到的東西了
光學顯微鏡加螢光劑,還是屬於物理學.